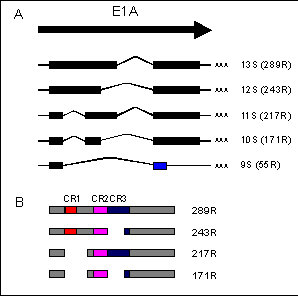
Figure 1: Structure of the E1A mRNA transcripts and postions of the conserved regions in the E1A proteins
A: Coding regions are represented as boxes and are all read in the same frame except for the second exon of the 9S mRNA (blue box)
B: Regions of conserved amino acid sequence in the E1A proteins are represented as CR1, CR2 and CR3.
A comparison of E1A sequences of various human and simian adenovirus serotypes has identified three regions of conserved amino acid homology (72,142). In Ad5, conserved region 1 (CR1) maps between amino acid 40-80, CR2 between amino acids 121-139, and CR3 between residues 140-188 which roughly coincides with the 13S unique region (Fig. 1B). Evolutionary conservation of these particular sequences suggests that they are critical for E1A function, but by no means limits the possibility that other regions are at least equally important.
The E1A proteins are proline rich, acidic, and localized in the nucleus. Rapid nuclear localization is mediated by a highly basic pentapeptide signal sequence (Lys-Arg-Pro-Arg-Pro) at the extreme carboxyl terminus of the polypeptides (84). The conformational constraints imposed by the high proline content likely limit the formation of substantial secondary structure in the E1A proteins. The extreme heat stability of bacterially produced E1A protein, which retains significant transcriptional activation activity even after boiling for five minutes (75), suggests that either E1A can readily refold to an active conformation, or that E1A can function as a random coil. The surprising ability of E1A protein to tolerate large deletions and insertions without total disruption of its biological activities, has led to the concept that E1A is a series of small modular domains that are relatively independent of surrounding sequences.
A potential metal binding domain with a consensus zinc finger motif (Cys-Xaa2-Cys-Xaa13-Cys-Xaa2-Cys) has been identified within the unique region of the E1A 289R protein. The larger E1A protein does bind a single zinc ion, and as expected the smaller E1A protein that lacks this region does not. This structure appears essential for transcriptional activation by E1A as substitution of glycine for any of the four cysteines within this motif not only abolishes the ability of E1A to bind Zn2+ but also results in a loss of transactivation activity (23). It is not known whether this metal binding structure mediates an interaction between E1A and DNA or with other proteins.
Post-translational modification of the E1A products is limited to phosphorylation that occurs at serine residues 89, 132, 219, and possibly 96 and 231 (30,31,134). The available evidence suggests that phosphorylation does little to regulate E1A activity (30,133).
Surprisingly, E1A can also function as an anti-oncogene to suppress transformation, metastasis and tumorigenicity. In addition, E1A can promote differentiation, partially reversing the transformed phenotype of cells, induce apoptosis and susceptibility to tumour necrosis factor and enhance susceptibility to the host cellular immune response (22,44,96).
E1A is also known to associate specifically with a number of cellular proteins. A large amount of work using E1A mutants has established which regions of E1A are necessary for these activities (see below)
The following sections provide brief introduction to a number of the E1A functions listed above. Numerous reviews cover each of these activities in far greater detail.
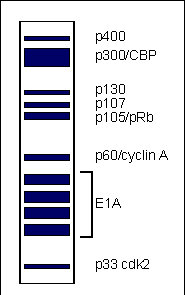
Figure 2: Depiction of an idealized fluorograph of the proteins co-immunoprecipitated with E1A from lysates of radiolabelled Ad5 infected cells
The regions of E1A required for association with most of these proteins have been mapped in a variety of cells using a multitude of mutants (Fig. 3B).
It seems likely that E1A functions indirectly through its association with these and other as yet unidentified cellular proteins. Our current assumption is that E1A binds to these proteins and either sequesters them, preventing their action, or modifies their activities.
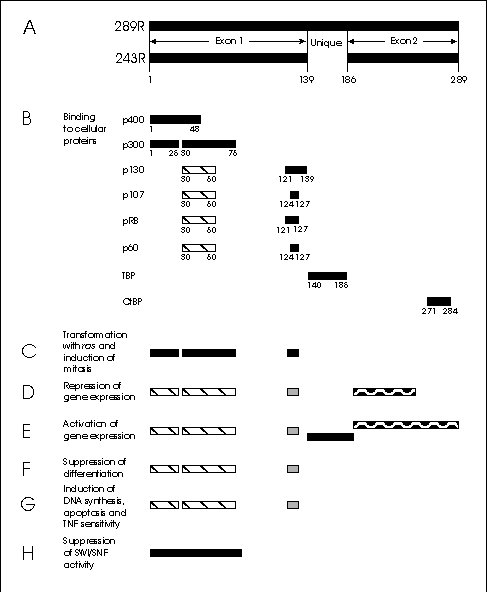
Figure 3: Map of the major E1A proteins and the regions required for selected E1A activities.
A) The 289 and 243 residue E1A proteins.
B) Consensus sites for binding to cellular proteins. The hatched regions are of secondary importance.
C-H: Regions required for:
C) Transformation with activated ras and induction of mitosis in BRK cells.
D) Repression of gene expression. Generally the hatched regions are necessary, but for repression of some genes either of the other two regions are required as well.
E) Activation of gene expression. The different shadings represent regions required for different pathways.
F) Suppression of differentiation. Depending on the cells, the minimum requirements are the hatched regions; either the hatched or the stippled region; or all three.
G) Induction of DNA synthesis, apoptosis and TNF sensitivity
H) Suppression of SWI/SNF function in Saccharomyces cerevisiae.
The E1A proteins alone are sufficient to immortalize primary rodent epithelial cells allowing them to proliferate indefinitely (62,94). These cells will not grow to high saturation densities, and do not exhibit the morphological characteristics of fully transformed cells unless E1A is expressed at very high levels (115). Mutational analysis has shown that regions in the N-terminal half of E1A, similar to those required for transformation, are also necessary for immortalization of primary BRK cells (106,114,127). Immortalization also requires a region near the C-terminus of E1A (106,128). Since the C-terminal half of E1A is not required for transformation with ras (144,156) or E1B (5), this suggests that whatever function resides in the C-terminus is provided by the cooperating oncogene.
CR3 consists of two domains (49,150). At its C-terminal end, residues 183-188 locate the E1A protein on a promoter by interacting with a promoter-bound transcription factor, in this way recruiting E1A to a promoter (103). Once recruited to a promoter, the activation domain in the N-terminal part of CR3, residues 141-178, containing a zinc finger (23,85,150), binds to TBP and activates transcription by stimulating the formation of a multiprotein transcription initiation complex with RNA polymerase II (15,48,61). Evidence suggests that the region of CR3 that binds TBP binds another cellular factor as well (15,48). Furthermore, trans-activation of some promoters requires one of two auxiliary elements in exon 2 besides CR3 (11).
A number of pathways have now been identified by which E1A exon 1 can induce gene expression (52,69,74,97,140,147). Of these, the most studied is the action of E1A in effectively freeing the E2F family of transcription factors from association with pRb, p107 and p130 (see Fig. 4). By freeing E2F, E1A activates transcription from a variety of genes containing E2F-binding sites in their promoters, including the adenovirus E2 early promoter (4,19,73,109), c-myc (60,131) and dihydrofolate reductase (59). Similar increases have been reported for the transforming oncogenes of other DNA tumour viruses that bind pRb, including HPV 16 E7 protein (104,108), and possibly the immediate early protein of human cytomegalovirus (146). In addition, HPV E7 protein and SV40 large T antigen disrupt complexes of E2F with pRb and cyclin A in vitro (18,102). Interestingly, E2F is not required for transcription of viral genes in SV40 and HPV, as it is in adenovirus. Instead, the release of E2F by these viral proteins appears to be related to the presence of E2F sites in the promoters of several S-phase specific cellular genes, activation of which probably facilitates viral growth (65).
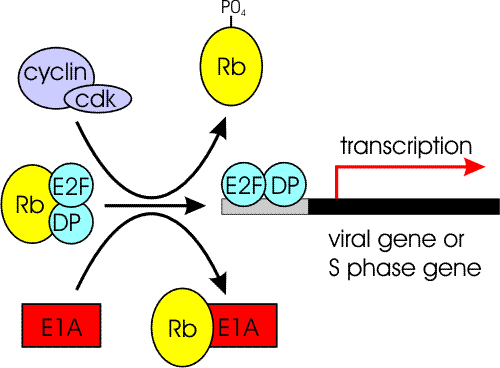
Figure 4: Effect of E1A on pRb/E2F function
In the G0/G1 phase of the cell cycle, hypo-phosphorylated pRb complexes with transcription factors of the E2F family (indicated as E2F and DP) preventing their ability to activate transcription. Cell cycle dependent phosphorylation of pRb by cyclin/cdk complexes releases E2F to activate transcription of target genes required for S-phase of the cell cycle. Expression of E1a overrides the normal cellular control of the pRb-E2F interaction by binding hypo-phosphorylated pRb and freeing E2F.
In primary BRK cells, which are regularly used for studies of transformation by E1A, the 243R protein activates expression of a range of viral early and late genes that lacked E2F binding sites in their promoters (97). Expression of viral late genes requires E1A to bind to p300. On the other hand, viral early genes and the cellular gene for PCNA are activated through at least two independent, redundant pathways, requiring binding to p300 and to pRb, respectively, as only E1A mutants that fail to bind to p300 and pRb simultaneously are defective (71,97). These results show that E1A can activate expression by binding to pRb through some mechanism that does not involve E2F. Besides activating gene expression directly, the p300/CBP pathway may also be able to activate the pRb/E2F pathway indirectly. E1A induces synthesis of p34cdc2 kinase in BRK cells through redundant p300/CBP and pRb pathways (27,147), indicating that binding to pRb is not essential for induction. Increased synthesis of p34cdc2 leads to increased phosphorylation of pRb (147). As only hypophosphorylated pRb complexes with and inactivates E2F (19), hyperphosphorylation likely results in the release of E2F and activation of E2F dependent transcription.
E1A also activates gene expression through the AP-1 family of transcription factors. These proteins and those of the similar ATF/CREB family contain a basic DNA-binding domain, a so-called leucine zipper, and an activation domain. Through their leucine zippers, the proteins form dimers either with themselves or with other members of the AP-1 and ATF families, and by binding to specific promoter sites, activate transcription. The sites to which the dimers bind are promoter elements that confer response to the phorbol ester TPA or to cyclic AMP (cAMP). The promoter of the gene c-jun binds a heterodimer of its own product, c-Jun, with ATF-2 (139), and E1A is able to stimulate transcription of this gene (139,140), apparently by causing hyperphosphorylation of the trans-activating domain of c-Jun (52). This action of E1A depends on CR1 but not on CR2 (140), which suggests that binding to p300/CBP is involved (52). In cooperation with cAMP, E1A also induces expression of c-fos (95), which is another member of the AP-1 family. This induction requires E1A to bind to p300 (46). In in vitro experiments, E1A, acting either on its own (24) or in cooperation with cAMP (46), increases binding of AP-1 to a TPA-responsive element; in the latter case, the increase requires E1A bind to p300/CBP (46). The influence of E1A on AP-1 activity provides a mechanism by which E1A may interact with signal transduction pathways in the cell.
Mutational analysis of E1A has mapped the domains of E1A required for enhancer repression primarily to a N-terminal region including CR1 (5,67,110,124,144)(Fig. 3D). This region correlates well with the region of E1A required for binding to p300/CBP (67,124), suggesting that transrepression is mediated through the association of E1A with p300/CBP. Several studies have also suggested a role for CR2 in transrepression (80,81,114), but this is not usually observed. As p300 and CBP act as transcriptional co-activators for a number of transcription factors (66), binding by E1A may sequester them making them unavailable for transcriptional activation. p300/CBP associates with another cellular protein referred to as p/CAF that is a histone acetylase and E1A can disrupt this interaction (158). Recruitment of p/CAF to a promoter by p300/CBP could result in promoter specific histone acetylation that helps overcome the repressive effects of assembly of genes as chromatin. Interestingly, in Saccharomyces cerevisiae , an N-terminal region of E1A suppresses SWI/SNF activation of gene expression (90), suggesting that E1A can repress transcription by blocking chromatin remodelling.(Fig. 3H)
E1A mutants that fail to repress also fail to transform (67,124), suggesting a requirement for transcriptional repression in oncogenic transformation. Although others have reported mutants that are defective for transrepression that still transform (5,76,144),at least some of these mutants were later found to repress transcription from a different enhancer (5,124), suggesting a dependence on the promoter.
In contrast to the cell lines described above, in undifferentiated mouse F9 teratocarcinoma cells E1A induces changes in morphology and gene expression characteristic of differentiation (91,100,145). Similar effects have been observed upon introduction of E1A into P19 embryonal carcinoma cells (118). Thus, in embryonal and teratocarcinoma cells, E1A can act as a tumour-suppressor to induce differentiation characteristics. Mapping studies indicate that induction of differentiation of P19 cells by E1A requires the N-terminal 25 residues and CR1 region and this correlates with binding to p300 (118). Whether this same region is required for induction of differentiation in F9 cells has not been determined.
It has been noted that introduction of E1A into a number of human tumour cell lines induces a conversion to a less refractile "flat" morphology, a reorganization of the cytoskeleton, a restoration of contact inhibition and a reduction in anchorage independent growth indicative of a less transformed, hence more highly differentiated state (42). It was later shown that expression of the 243R E1A protein induces the expression of epithelial characteristics in a diverse array of human tumour cells regardless of parental cell type (43). This surprising result has led to speculation that the epithelial cell phenotype may be a "default" that requires only "ubiquitous" transcription factors. Thus, the mechanism by which E1A stimulates conversion to the epithelial phenotype may result from its ability to inhibit the expression or activity of cell type specific transcription factors (43). If this were indeed the case, the regions of E1A involved in suppression of differentiation would likely correlate with those involved in induction of the epithelial phenotype, but this has yet to be determined.
The regions of the 243R protein required for induction of apoptosis are the same as those required for inducing quiescent cells to enter and traverse the cell cycle (99) (Fig. 3G). Surprisingly, the 243R protein induces apoptosis only in cells prevented from proliferating by cell-cell contact or by serum deprivation (99,107), suggesting that induction of apoptosis is an unintended consequence of a proliferation block opposing induction of cell division by E1A (99). A similar conclusion has been made for the induction of apoptosis by inappropriate expression of c-myc protein (39). To overcome the tendency of E1A to induce apoptosis, the viral E1B encoded 19 kDa and 55 kDa products suppress apoptosis (107) and this may explain the co-operativity between E1A and E1B in cell transformation.
In addition to inducing apoptosis, E1A can also sensitize cells to killing by tumour necrosis factor (TNF) (20,28). TNF was originally identified through its anti-tumour activity (16) and can induce both necrotic and apoptotic forms of cell lysis in transformed cells (77). Detailed analysis of the regions of E1A responsible for induction of TNF sensitivity shows an exact correlation with those regions required for induction of DNA synthesis (117) (Fig. 3G). These results are largely consistent with earlier less comprehensive studies (1,29,137). In infected cells, the viral E3 encoded 10.4 kDa, 14.5 kDa and 14.7 kDa proteins suppress TNF mediated cytotoxicity induced by E1A (50,51).
In a semi-permissive infection of rodent cells with human adenovirus, E1A appears to perform its proper functions in preparing the host cell for maximal viral replication. The E1A proteins bind to cellular regulatory proteins such as pRb and p300, alter gene expression and force the infected cell to enter the cell cycle. In many of these cells the life cycle of human adenovirus is not properly completed, allowing some cells to survive. As the result of programming the infected cell to commence division, E1A apparently eliminates the option for these cells to undergo differentiation, the opposite process to cell division. The extended proliferative capacity induced by E1A in these cells probably allows the accumulation of other genetic aberrations, leading to the transformed phenotype. This argument is firmly supported by the requirement for identical regions of E1A for both transformation, and entry and passage through the cell cycle.
Interestingly, in the absence of E1B, the presence of E1A induces programmed cell death or apoptosis in otherwise healthy cells. Induction of apoptosis appears to require exactly the same regions of E1A necessary for induction of DNA synthesis, suggesting that the same signals produced by E1A that induce cell division can trigger suicide (99). This undesirable side effect would hamper efficient virus production. The virus appears to have compensated by evolving the E1B 19 kDa protein, which can inhibit apoptosis by an as yet unknown mechanism. It has been speculated that this ability to block apoptosis may be one reason why E1B 19 kDa can cooperate with E1A to transform cells (107). This is a definite possibility as the product of the proto-oncogene Bcl-2, which is known to block apoptosis, but is otherwise distinct from the E1B 19 kDa protein in structure and location, can also cooperate with E1A to transform primary BRK cells (107). This suggests that other oncogenes, such as activated ras, that can cooperate with E1A to transform cells might function to block apoptosis as well.
Thus, E1A affects each of the three processes, division, differentiation and death, that a hypothetical cell sitting in the Go phase of the cell cycle could choose (or be forced) into. Significantly, the ability of E1A to do this depends on two key domains that are required for binding to pRb and p300/CBP. These studies demonstrate that the interaction and likely inactivation of pRb and p300/CBP by E1A also appears central to the ability of E1A to reprogram gene expression (6), strengthening the argument that it is the changes in gene expression induced by E1A that ultimately result in the effects E1A has on cell growth and development.
Without question, far more must be learned about how the interactions of E1A with cellular factors alter gene expression before we understand in detail how E1A alters cell growth and leads to transformation. Nevertheless the recognition that E1A acts to regulate gene expression through multiple distinct mechanisms and the apparent connection of some of these with transformation is significant.