Research in my laboratory is centred on interrelated projects that each intersects with the molecular themes of cell signaling and proliferative control, chromatin organization, and genome integrity.



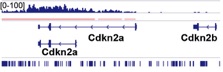
TUNEL staining of irradiated tissue.
Rb1 mutant mice.
Telomere staining of chromosomes.
Anaphase bridge in lymphoma.
ChIP-seq data.
Alazarin Red stain of DREAM mutant mouse skeleton.
Loss of cell cycle control is one of the defining features of cancer, as uncontrolled cell division fuels tumor growth. My laboratory has had a long standing interest in molecular mechanisms of proliferative control and in linking their misregulation to cancer progression and therapeutic response. Using an allelic series of gene targeted mice where each possess a different point mutation in the cell cycle regulating RB gene we have discovered that proliferative control is not easily defined by single biochemical functions. Our data indicates that interrelated networks controlling E2F transcription and cyclin dependent kinase activity coordinate cell cycle arrest events that depend on the RB tumor suppressor protein. With the emergence of Palbociclib and other agents that effectively 'turn on' RB cell cycle arrest function in cancer, we expect that the competence of the entire RB dependent cell cycle arrest network will play a key role in determining efficacy of these drugs. In addition, acquired resistance to targeted agents is common and this further suggests that loss of components of the RB cell cycle arrest network will contribute to the effectiveness of these drugs. Defining components and understanding their relationships with one another in this RB-dependent proliferative control and drug response network will be important to treating hormone dependent breast cancer in the future.
Numerous proteins function to organize mammalian chromatin and compact it into mitotic chromosomes during mitosis. Errors in organizing chromatin structure inevitably lead to genome instability through DNA breaks, abnormal DNA replication, and errors in mitosis. While this contributes to cancer pathogenesis in some contexts, it is also a potential target for cancer treatment as many chemotherapeutics exacerbate genome instability to cause cell death. In addition, loss of heterochromatin can misexpress silenced RNA sequences that in turn stimulate an immune response and potentially contribute to mechanisms of cancer immune therapy. Work in this area is focused mainly on Condensin II and EZH2, two chromatin structure regulators that are recruited to DNA by the RB-tumor suppessor through a cell cycle independent interaction with E2F1. These projects seek to understand the fundamental organization mechanisms of heterochromatin and how they are altered in cancer, or by small molecule inhibitors. Ultimately, the relevance of these chromatin organizing mechanisms are investigated using gene-targeted mouse models that are deficient for heterochromatin assembly. We expect this work will point to new vulnerabilities in cancer and better describe the effects of chromatin altering therapeutics that are currently in clinical trials. This will be particularly important in osteosarcoma, prostate, and lung cancer treatment in the future.
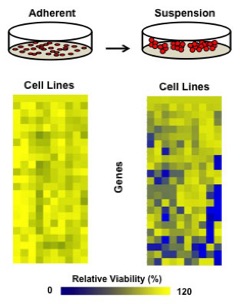
Cancer is most often characterized by inappropriate cell proliferation. Work in this area of my lab's research is focused on circumstances where cancer cells ironically stop growing. The ability of cancer cells to halt proliferation is critical for their ability to survive under stressful conditions where oxygen and nutrients are limiting. In addition, prolonged cell cycle arrest allows cancer cells to escape the toxic effects of most chemotherapeutic agents that take advantage of the vulnerabilities of rapidly dividing cells. These 'dormant' cancer cells are a significant source of disease relapse for cancer survivors, underscoring the importance of understanding how cancer cells can escape from a rapidly proliferating state and how they survive. We are using systematic approaches to investigate which genes are needed for cancer cells to arrest under stressful conditions. In addition, we are studying the normal physiological roles for these arrest mechanisms in gene targeted mouse models to understand their normal role in mammalian physiology. One contributor to cell dormancy is the DREAM complex and we are studying its regulation as well as mutant mice that are defective for assembling this complex. Understanding and exploiting the vulnerabilities of dormant cancer cells will be key to extending the lives of breast and ovarian cancer survivors.
Integration of proliferative control mechanisms by the RB protein in cancer progression and therapy.
Roles of chromatin structure in genome stability and inflammation.
Negative growth control and survival mechanisms in cancer cell dormancy.
We use a range of methods from in vitro biochemical experiments and cell culture, to gene targeted mouse models of disease. We also use the latest CRISPR based methods and next generation sequencing to ensure our experiments are systematic and comprehensive.